Peptoids
Peptoids, or N-substituted glycines, are peptidomimetics with chemistry similar to proteins, which, at the same time, remove their functional limitations. Structurally, peptides have their defining side-chains attached to the a-carbon in the backbone, leaving a hydrogen atom attached to the nitrogen atom. This means that the formation of the peptide bond is almost always a trans isomer (due to steric clash) and leads to the possibility of hydrogen bonding. Conversely, in peptoids, the side-chains are attached to the backbone nitrogen (Figure 1). This introduces the possibility of forming both cis and trans isomers for the peptide bond. Furthermore, since there is no hydrogen atom attached to an electronegative atom, there is no possibility of hydrogen bonding (although it can be introduced depending on the nature of the side-chain) [1-7].
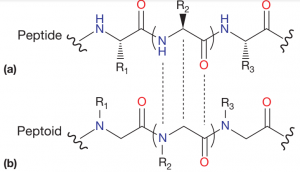
Structural comparison between peptides and peptoids. Adapted from Seo et. al. [7]
Heteropolymers with biologically inspired designs are in huge demand in industry for their sequence specificity, ability to act as catalysts for a plethora of reactions and their functionalities [2,3]. This has led to an increase in the research and development of biomolecules such as proteins and peptoids. One of the major limitations of proteins is their degeneration in the presence of proteases, low thermal tolerance and susceptibility to pH change. This occurs largely due to disruptions in the hydrogen bonds of the backbone chain, which control the secondary and tertiary protein structures. On the other hand, for peptoids, secondary structure formation is usually dictated by steric interactions, which is not as susceptible to external conditions. This makes peptoids resistant to thermal and chemical denaturation. Furthermore, peptoids are able to exhibit all the secondary and tertiary structures seen by peptides, as well as some of the structures not seen before [5,6]. This opens new avenues for applications not previously explored for peptide-like biomaterials. Another interesting biological application is that peptoids are much more capable of penetrating the cell membrane and interacting with the cytoplasm and nuclei, as opposed to peptides [3].
Therefore, peptoids represent a peptide-like class of biologically compatible materials that can be used in a wide variety of applications such as drug carriers and catalysts in biochemical reactions. Due to the enhanced cell penetration of peptoids as opposed to peptides, they also find interesting applications as an intracellular delivery vehicle for nucleic acids [3]. Peptoids also have the ability to form biofilms and therefore can be used as effective antifouling agents [7].
Peptoids and Simulations
Peptoids are synthetic materials that were first developed in the 1980s [1]. Even though they are fairly new on the “science timescale”, there has been huge progress in their synthesis methods. In contrast, the development of simulation methods for these systems has fallen short by a huge margin [4]. The main reason for this shortcoming is the fact that unlike peptides, peptoids have to sample both the cis- and trans- forms to find the exact energy minima of the system, which is often difficult to model. Modifying the existing peptide models to use for peptoids has allowed the prediction of backbone conformations to a certain degree of accuracy, but they are not specific to peptoids and, as mentioned above, are not able to sample the complete peptoid energy landscape.
Some attempts have been made for ab-initio studies of small peptoid molecules [18]. Ab-initio methods depend completely on quantum mechanical structure of the molecule which makes it highly accurate, but this takes up much more computational time than other methods, which is their one major drawback. This makes quantum calculations the bottleneck step for using ab-initio to study peptoids. They have largely been used for predicting the optimized peptoid monomer backbone structure which, in turn, supports all-atom force fields, the most common methods to study peptoids. An interesting approach would be to use a combination of all-atom and ab-initio methods [19]. The major sampling of the energy landscape for a particular application would be done by all atom force fields, which would give a potential minimum. Being the crude model that it is, fine-tuning of the structure would next be done using ab-initio methods. This might even enable us to consider significantly longer chains in ab-initio simulations. This technique has demonstrated its usefulness by helping predict the secondary structure observed in a class of peptoids, known as peptoid ribbons [20]

Sarcosine, the simplest peptoid

A peptoid with N-alkoxy linkage
One of the first forcefields developed specifically for peptoids was MFTOID (Molecular Foundry PepTOID) which used an additive CHARMM-like force field, with parameters fitted specifically to peptoids [21]. Since they used CHARMM as their starting point, they also saw overstabilization of secondary structure. Furthermore, they fitted the backbone parameters to sarcosine (Fig. 4a), which raises the issue of transferability of potential parameters. Similar to peptide studies, we should be able to extend the parameters to simple peptoids. Another approach might be needed for molecules which have the defining side chain not attached by a methyl linkage to the backbone, such as Fig. 4b, where we see an alkoxy linkage.Most peptoid modeling studies employ all-atom force fields, which can be used to simulate a large molecule without using too much computational time. A probable limitation can be the incorrect energetics followed by the simulations, which is not really a concern in cases where one just seeks to study the equilibrium structures. They can also provide useful details about the assembly process for shorter peptoid chains which join together to form longer peptoid chains. Most of the methods focus on developing a forcefield for the molecules using either smaller scale simulations such as ab-initio, known as bottom-up coarse graining or fitting forcefield parameters to available experimental data and results, known as top-down coarse graining. Several initial attempts used protein force fields such as CHARMM and AMBER to predict minimum energy structures in vacuum or in a solvent for simple peptoids such as sarcosine [19, 21-23]. Some studies led to overstabilization of the secondary structures whereas others gave wrong results for the dihedral angles.
Another attempt was made by Weiser et. al. [22] who used the general formulation of CHARMM developed for complicated drug-like molecules, in addition to the already included molecule types in CHARMM. The new atom type introduced, NTOID, focused on refitting new parameters to the existing model except for the nitrogen atom in the backbone. All the parameters involving the peptoid nitrogen were recalculated as all the major differences between peptides and peptoids are focused around the nitrogen atom. In certain cases, especially those with much bulkier side chains, where there are larger gaps in energies between configurations, methods different than those for peptides might be needed to scale the complete energy landscape. These researchers were able to generate Ramachandran plots accurately for sample peptoid chains, reproducing both the cis and trans confirmations along the way.